Dr. Adriel Dominguez Garcia

Fields of interest | TD-DFT/B excitons in TiO2 Real-time electron dynamics in photocatalysis |
| Publications | Google Scholar |
Research
My main research focus is on the structural, electronic and optical properties of semiconductor materials and molecules, as well as their combination to form a superior functional complex, by means of atomistic simulations based on quantum mechanical (QM) approaches. Due to my interest in extended compound systems and long simulation time frames, I focus on Density Functional Theory (DFT) and approximate DFT formalisms as main theoretical tools, which provide a good compromise between numerical accuracy and computational cost. I however employ a wide spectrum of QM methods in my research, ranging from Hartree-Fock to many body perturbation theory (MBPT).
Optical Properties of TiO2 nanoparticles and single crystals

Titania (TiO2) is widely employed in fields like catalysis, photocatalysis and light-energy conversion. A deep understanding of this material may lead to important advances in the air purification and renewable energy segments, which are two high priority fields for scientists. Especial efforts have therefore been put in the in-depth study of TiO2. However, many aspects of it remain elusive and the use of sophisticated methodologists applied to its study continues to be a challenge. We perform first principle calculations supported by state-of-art spectroscopy measurements to study the excitonic properties of titania, with special emphasis on the interplay between the fundamental charge excitations and its lattice degrees of freedom. We work towards the improvement of the theoretical description of the electron-phonon interaction in the material, including many body effects. We also study the mobility and transport of the charge carriers along different directions inside the crystal.
Simulations of visible- and UV-light driven reactions on TiO2 surfaces
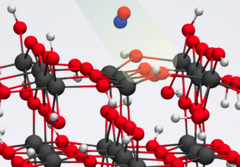
TiO2 has immense potential in the fight against environmental pollution, as it has been shown to be a powerful and suitable photocatalyst. We focus on the photocatalytic nature of different TiO2 facets, especially for air purification applications. We investigate the mechanisms and pathways of the different photoreactions taking place at the surfaces by means of linear response TD-DFTB and Ehrenfest molecular dynamics simulations. For instance, we study how the presence of water and oxygen affects the degradation of NO at the surfaces of the semiconductor material. We also investigate the photooxidation of CO to CO2 and the role played by oxygen molecules in this photoreaction.
Electronic and optical properties of 2D materials and van der Waals heterostructures
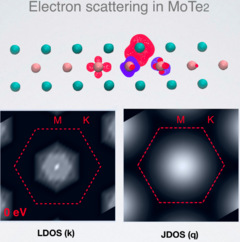
Atomically thin materials like graphene and transition metal dichalcogenides (TMD) have unique physical properties. The vertical stacking of these materials and III-V binary compounds via van der Waals (vdW) forces has lead to the synthesis and prediction of novel heterostructures with a wide spectrum of electronic and optical characteristics. We investigate new vdW layered heterostructures using many body perturbation theory and DFT. We have focused on the excitonic states of recently synthesised materials as well as proposed heterostructures combining InSe with graphene, germanene and antimonene. We also study phonon-assisted excitations in WSe2 and the electronic scattering in defective MoTe2.
Method Development in Density Functional Theory

DFT approaches are an efficient alternative to wavefunction-based methods. The Density Functional Tight Binding (DFTB) approximation and its time-dependent extension, TD-DFTB, combine the accuracy of ab initio DFT with the efficiency of semiempirical methods. We work on the extension and sophistication of methods within DFTB and integrate them into the DFTB+ code package. For example, we have designed and implemented a simplified particle-particle random phase approximation (pp-RPA) scheme which is orders of magnitude faster than the original formalism, while its numerical accuracy is retained. Currently, we are developing a localised orbital scaling correction within DFTB, which seeks to reduce the delocalisation error of the electronic states.