Dr. Zhen Tong

| Fields of interest | Thermal transport in thermoelectric material |
| Publications | Google Scholar |
| Personal Homepage |
Research
Research focuses on the fundamental understanding of thermophysical properties and engineering the properties to enhance energy conversion and utilization efficiency. At the fundamental level, I develop and implement numerical simulation methods for thermal transport properties (especially thermal conductivity and thermal emissivity) at different scales. Our target is to develop a fundamental understanding of heat conduction and radiation phenomena in a variety of materials, including solid materials and composite materials. The energy conversion and utilization research is targeted at enhanced energy conversion efficiency and efficient thermal energy dissipation by tuning the thermal properties of materials. The application includes solar-thermal energy conversion, radiative cooling, etc.
High efficiency solar-thermal energy conversion system design
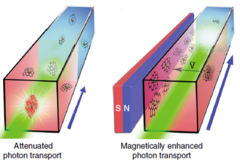
Currently, solar-thermal energy storage within phase-change materials relies on adding high thermal-conductivity fillers to improve the thermal-diffusion-based charging rate, which often leads to limited enhancement of charging speed and sacrificed energy storage capacity. Here we report the exploration of a magnetically enhanced photon-transport-based charging approach, which enables the dynamic tuning of the distribution of optical absorbers dispersed within phase-change materials, to simultaneously achieve fast charging rates, large phase change enthalpy, and high solar-thermal energy conversion efficiency. Compared with conventional thermal charging, the optical charging strategy improves the charging rate by more than 270% and triples the amount of overall stored thermal energy. This superior performance results from the distinct step-by-step photon-transport charging mechanism and the increased latent heat storage through magnetic manipulation of the dynamic distribution of optical absorbers.
Predict thermal conductivity by including phonon-phonon and electron-phonon scatterings from first-principles
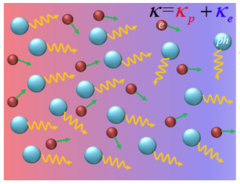
Separating electron and phonon components in thermal conductivity is imperative for understanding thermal transport in metals and highly desirable in many applications. In this work, we predict the mode-dependent electron and phonon thermal conductivities of 18 different metals at room temperature from first principles. Our first-principles predictions, in general, agree well with those available experimental data. For phonon thermal conductivity, we find that it is in the range of 2–18 W/mK, which accounts for 1%–40% of the total thermal conductivity. It is also found that the phonon thermal conductivities in transition metals and transition-intermetallic compounds (TICs) are non-negligible compared to noble metals due to the high phonon group velocities of the former. We further show that the electron-phonon coupling effect on phonon thermal conductivity in transition metals and intermetallic compounds is stronger than that of nobles, which is attributed to the larger electron-phonon coupling constant with a high electron density of states within the Fermi window and high phonon frequency in the former. For electron thermal conductivity, we observe that the transition metals and TICs have lower electron thermal conductivities compared to noble metals.
Prediction of thermal emissivity by including three and four-phonon scatterings from first-principles
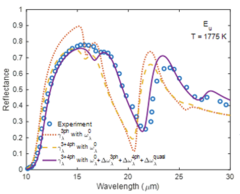
Recently, first-principles calculations based on density functional theory have been widely used to predict the temperature-dependent infrared spectrum of polar materials, but the calculations are usually limited to the harmonic frequency (0 K) and three-phonon scattering damping for the zone-center infrared-active optical phonon modes, and fail to predict the high-temperature infrared optical properties of materials such as sapphire (α-Al2O3), GaAs, TiO2, etc., due to the neglect of high-order phonon scattering damping and phonon frequency shift. In this work, we implemented first-principles calculations to predict the temperature-dependent infrared dielectric function of polar materials by including four-phonon scattering and phonon frequency shift. The temperature-dependent phonon damping by including three- and four-phonon scattering as well as the phonon frequency shift by including cubic and quartic anharmonicity and the thermal expansion effect are calculated based on anharmonic lattice dynamics method. The infrared dielectric function of α-Al2O3 is parameterized, and then the temperature-dependent infrared optical reflectance is determined. We find that our predictions agree better with the experimental data.