Xiangyu Guo

Fields of interest | 2D materials, Atomically dispersed catalysts Ferroelectrics; electro- and photo-catalysis. |
| Publications | Google Scholar |
Research
The research training and experience lie in the area of atomic modelling and simulation for materials via density functional theory computations. I have been working on the development of a universal principle to understand the catalytic limitations of the materials and for a rational design of high-performance catalysts. The ultimate aim of the research is to identify the stable and efficient catalysts to tackle the activity and selectivity challenges toward the production of future fuels, such as H2, NH3, CH4, and CH3OH, etc.
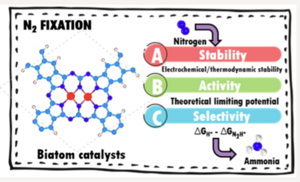
Developing efficient catalysts for nitrogen fixation is becoming increasingly important but is still challenging due to the lack of robust design criteria for tackling the activity and selectivity problems, especially for electrochemical nitrogen reduction reaction (NRR). Herein, by means of large-scale density functional theory (DFT) computations, we reported a descriptor-based design principle to explore the large composition space of two-dimensional (2D) biatom catalysts (BACs), namely, metal dimers supported on 2D expanded phthalocyanine (M2-Pc or MM′-Pc), toward the NRR at the acid conditions. We sampled both homonuclear (M2-Pc) and heteronuclear (MM′-Pc) BACs and constructed the activity map of BACs by using N2H* adsorption energy as the activity descriptor, which reduces the number of promising catalyst candidates from over 900 to less than 100. This strategy allowed us to readily identify 3 homonuclear and 28 heteronuclear BACs, which could break the metal-based activity benchmark toward the efficient NRR.
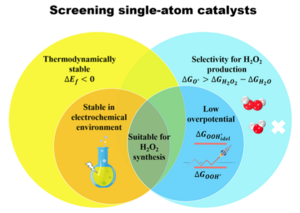
Herein, by means of density functional theory (DFT) computations, we reported the feasibility of several classes of important and representative experimentally achievable single-atom catalysts (SACs) toward two-electron ORR, paying attention to their stability, selectivity, and activity at the acidic medium. Starting from 210 two-dimensional (2D) SACs, we demonstrated that 31 SACs have the potential to break the metal-based scaling relations and simultaneously achieve high activity and selectivity toward H2O2 production and screened out 7 SACs with higher activity than the PtHg4 in acidic media. Especially, a noble metal-free SAC, namely, a single Zn atom centered phthalocyanine (Zn@Pc-N4), has a remarkable activity improvement with a small overpotential of 0.15 V. Moreover, using multivariable analysis and machine-learning techniques, we provided a comprehensive understanding of the underlying origin of the selectivity and activity of SACs and unveiled the intrinsic correlations between structure and catalytic performance. This work may pave a way to the design and discovery of more promising materials for H2O2 production.

To achieve efficient ammonia synthesis via electrochemical nitrogen reduction reaction (NRR), a qualified catalyst should have both high specific activity and large active surface area. However, integrating these two merits into one single material remains a big challenge due to the difficulty in balancing multiple reaction intermediates. Here, it is demonstrated that the boron-analogues of MXenes, namely “MBenes”, could cope with the challenge and achieve the high activity and large reaction region simultaneously toward NRR. Using extensive density functional theory computations and taking 16 MBenes as representatives, it is identified that seven MBenes (CrB, MoB, WB, Mo2B, V3B4, CrMnB2, and CrFeB2) not only have intrinsic basal plane activity for NRR with limiting potentials ranging from −0.22 to −0.82 V, but also possess superior capability of suppressing the competitive hydrogen evolution reaction.