Dr. Adam McSloy

| Fields of interest | Machine learning, solid state Ionics, and electronic structure methods |
| Publications | Google Scholar |
Research
I work in the field of computational and theoretical chemistry. In my research, I aim to explore how machine learning methods can be used to augment and improve traditional computational chemistry simulation methods, chiefly by reducing computational complexity and increasing flexibility. Currently, I am primarily working on developing a python-based machine learning framework for tight binding theory type methods, such as “DFTB”.
A deep neural network for molecular wave functions in quasi-atomic minimal basis representation
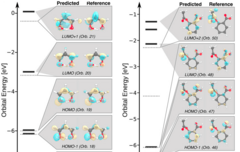
The emergence of machine learning methods in quantum chemistry provides new methods to revisit an old problem: Can the predictive accuracy of electronic structure calculations be decoupled from their numerical bottlenecks? Previous attempts to answer this question have, among other methods, given rise to semi-empirical quantum chemistry in minimal basis representation. We present an adaptation of the recently proposed SchNet for Orbitals (SchNOrb) deep convolutional neural network model [K. T. Schütt et al., Nat. Commun. 10, 5024 (2019)] for electronic wave functions in an optimized quasi-atomic minimal basis representation. For five organic molecules ranging from 5 to 13 heavy atoms, the model accurately predicts molecular orbital energies and wave functions and provides access to derived properties for chemical bonding analysis.
Structure and ion transport of lithium-rich Li1+xAlxTi2−x(PO4)3 with 0.3<x<0.5
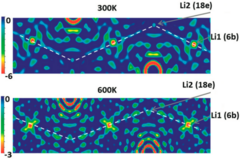
New solid-state electrolytes are becoming increasingly sought after in the drive to replace flammable liquid electrolytes. To this end, several Li conducting solids have been identified as promising candidates including Li stuffed garnets and more recently Li-rich materials such as Li1+xAlxTi2−x(PO4)3 with 0.3< x <0.5. However, the structure/property relationships of LATP are incredibly sensitive to synthesis conditions and therefore challenging to optimise. In this joint computational and experimental investigation, we examine the structural sensitivities by modelling the site occupancies at varying temperature, which clarifies previously reported discrepancies of the crystal structures. Furthermore, we investigate the Li ion transport properties which have not reported computationally before. We confirm from our simulations that the migration pathway only involves the M1(6b) and M2(18e) site, in excellent agreement with the neutron diffraction data, clarifying all past controversies regarding the Li ion occupancies in LATP. Interestingly, we calculate low migration barriers (0.3 eV) in line with experimental findings but also show evidence of Li ion trapping on Al doping in LATP (where x = 0.4), possibly explaining the experimental observation that the Li ion conductivity does not improve above x = 0.3, due to a stronger repulsion between Li+–>Ti4+ compared to Li+–>Al3+. Furthermore, our calculated ionic conductivities are in excellent agreement with experimental values, highlighting the robustness of our computational models.